Table of Contents >> Show >> Hide
- CJD vs. Mad Cow Disease: What’s the Difference?
- Prions: The “Germ” That Isn’t a Germ
- The Main Types of Creutzfeldt-Jakob Disease
- Symptoms: What CJD and vCJD Can Look Like
- Diagnosis: How Doctors Evaluate Suspected Prion Disease
- Transmission: What Spreads Prion Disease (and What Doesn’t)
- Mad Cow Disease (BSE): The Basics and Why U.S. Risk Is Low
- Food Safety Questions People Actually Ask
- Treatment: What Can Be Done?
- Healthcare Infection Control: Why Prion Protocols Exist
- Frequently Asked Questions (FAQ)
- Conclusion
- Experiences: What Patients, Families, and Clinicians Often Share (A 500-Word Perspective)
If you’ve ever heard someone whisper “mad cow disease” like it’s a horror movie title (it kind of is), you’re not alone.
The story gets even stranger when you learn the villain isn’t a virus, bacterium, or parasiteit’s a protein that folds wrong,
convinces other proteins to copy its terrible “origami,” and then wreaks havoc on the brain.
This article breaks down Creutzfeldt-Jakob disease (CJD), variant CJD (vCJD), and
mad cow disease (also called bovine spongiform encephalopathy, or BSE) in plain English.
We’ll cover what’s connected, what’s not, what symptoms look like, how doctors test for prion disease, and why U.S. food safeguards matter.
(Spoiler: the average American is far more likely to get struck by lightning than to get vCJDbut “far more likely” doesn’t mean “never,”
so it’s worth understanding the basics.)
CJD vs. Mad Cow Disease: What’s the Difference?
Let’s start with the quick translation:
-
CJD (Creutzfeldt-Jakob disease) is a human prion disease that causes rapidly progressive brain damage.
It’s rare and, tragically, almost always fatal. - BSE (bovine spongiform encephalopathy), nicknamed mad cow disease, is a prion disease in cattle.
-
vCJD (variant CJD) is the human illness that has been linked to eating beef from cows infected with BSE.
It’s different from “classic” CJD in how it tends to start, who it affects, and how long it lasts.
Think of it as a family of related disasters, all caused by prionsbut showing up in different species and in different ways.
Prions: The “Germ” That Isn’t a Germ
A prion is an abnormal version of a normal protein found in the body (especially the brain).
The abnormal form can trigger normal proteins to misfold toolike one bad domino knocking over a whole line.
Unlike most infectious agents, prions don’t have DNA or RNA. That means:
- Antibiotics don’t help (they target bacteria).
- Antivirals don’t help (they target viruses).
- The immune system often doesn’t “see” prions as a typical invader.
- Prions can be unusually resistant to routine disinfection methods.
As prions accumulate, brain tissue can develop microscopic holesgiving it a “spongy” appearance under the microscope.
That’s why these conditions are often grouped as transmissible spongiform encephalopathies (TSEs).
The Main Types of Creutzfeldt-Jakob Disease
“CJD” is often used as an umbrella term, but there are multiple forms. Understanding the type matters for risk assessment,
infection control, and family counseling.
Sporadic CJD (sCJD): The Most Common Form
Sporadic CJD happens without a known trigger. It’s the most common type of classic CJD.
Most cases occur in older adults, and symptoms progress quicklyoften over weeks to months.
Researchers believe sporadic CJD may arise when a normal prion protein misfolds “by accident” and then spreads that misfolding.
It’s nobody’s fault. It’s biology being unfair.
Genetic (Familial) CJD: When a Mutation Raises Risk
Some cases are caused by inherited changes in the PRNP gene, which helps code for prion protein.
In these families, prion disease risk can be passed down through generations.
Important nuance: a genetic predisposition doesn’t mean symptoms appear at the same age or in the same way for everyone.
Families facing this often benefit from genetic counseling and specialized neurology care.
Acquired (Iatrogenic) CJD: Rare, Medical-Related Transmission
A small number of CJD cases have historically been linked to specific medical exposuressuch as certain transplanted tissues
or older medical products derived from human tissue (for example, some past uses of cadaver-derived pituitary hormones).
Modern safeguards have made these events extremely rare, but the history matters because prions behave differently from typical pathogens.
Variant CJD (vCJD): The One Connected to “Mad Cow”
Variant CJD is the form linked to eating food contaminated with the BSE prion.
It was first identified in the 1990s during the BSE crisis abroad, and it has remained rare.
Compared with sporadic CJD, vCJD tends to:
- affect younger people more often,
- start with psychiatric or behavioral symptoms more commonly, and
- progress over a longer time frame (often around a year or a bit more).
Symptoms: What CJD and vCJD Can Look Like
Prion diseases can start subtly and then accelerate with terrifying speed. Symptoms vary, but there are patterns doctors watch for.
The biggest red flag is rapidly progressive dementiameaning a person’s thinking and function decline fast,
not over years.
Common Symptoms in Classic CJD
- Rapid cognitive decline: confusion, memory problems, impaired judgment
- Personality or behavior changes: anxiety, irritability, withdrawal
- Coordination problems: unsteady gait, clumsiness, dizziness
- Visual disturbances: blurred vision, visual field changes
- Myoclonus: sudden jerking movements (often later, but sometimes early)
- Speech and swallowing difficulties as the disease advances
Common Symptoms in Variant CJD
- Early psychiatric symptoms: depression, anxiety, mood changes
- Sensory symptoms: unusual sensations (like tingling or burning)
- Neurologic decline: coordination issues, cognitive impairment, movement problems
- Longer course compared with classic CJD
A Realistic Timeline Example (Not a Diagnosis)
Here’s the kind of pattern that raises clinical concern:
- Weeks 1–4: new confusion, personality shift, sleep changes, subtle balance problems
- Weeks 4–8: worsening memory and function, clear coordination issues, possible visual problems
- Months 2–6: rapid loss of independence, difficulty speaking/swallowing, abnormal movements
Many illnesses can cause rapid decline (some treatable), so doctors approach this as a “rule-out emergency,” not a label to slap on casually.
Diagnosis: How Doctors Evaluate Suspected Prion Disease
There’s no single office swab for CJD. Diagnosis usually involves a careful evaluation to:
(1) look for findings consistent with prion disease and (2) rule out other causes of rapidly progressive dementia that may be treatable.
Key Tests Commonly Used
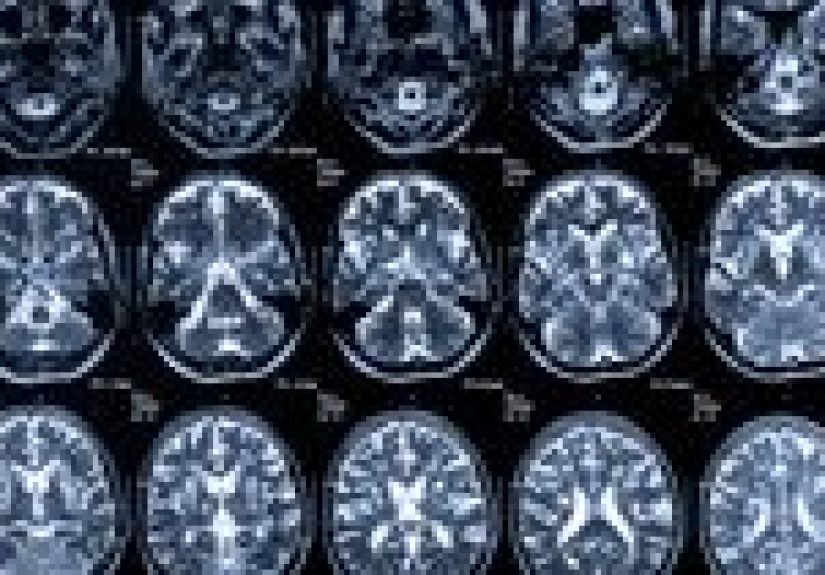
- Brain MRI (DWI/FLAIR sequences): often the most helpful imaging tool for classic CJD patterns.
- EEG: may show characteristic patterns in some cases, though it’s not definitive on its own.
-
Spinal fluid (CSF) tests:
- RT-QuIC (real-time quaking-induced conversion), which looks for evidence of abnormal prion protein activity
- supportive biomarkers such as 14-3-3 and total tau (not specific, but can support the picture)
- Genetic testing when familial prion disease is a concern.
- Additional bloodwork, imaging, and infectious/autoimmune tests to rule out mimics.
How vCJD Can Be Evaluated
vCJD has its own diagnostic criteria and may involve different supportive findings.
Definitive confirmation typically requires specialized testing of tissue.
Clinicians also consider travel and exposure history, given how rare vCJD is in the United States.
Confirming the Diagnosis
The only way to confirm prion disease with absolute certainty has traditionally been examining brain tissue,
typically after death. Because brain biopsy carries risks and often doesn’t change treatment, many cases are classified as
“probable” based on clinical features and test results.
In the U.S., specialized reference centers support state-of-the-art diagnostic testing and surveillance, helping clinicians and families
get answers as accurately and safely as possible.
Transmission: What Spreads Prion Disease (and What Doesn’t)
Let’s reduce panic first: classic CJD is not spread by casual contact. You cannot “catch” it by hugging someone,
sharing a meal, coughing near them, or sitting on the same couch (even if the couch is suspiciously old).
Prions are concentrated in certain tissuesparticularly the brain, spinal cord, and some nervous system tissues.
Transmission concerns historically include:
- specific medical exposures involving high-risk tissues (now heavily controlled),
- rare contamination scenarios involving instruments if not handled under prion protocols, and
- for vCJD, foodborne exposure to BSE-contaminated beef products (the reason food safety controls exist).
Mad Cow Disease (BSE): The Basics and Why U.S. Risk Is Low
BSE is a prion disease in cattle. It became infamous after outbreaks linked to feeding practices that recycled
infected animal protein back into cattle feedcreating a prion “echo chamber.”
In response, the United States built multiple, overlapping safeguards to reduce risk:
1) Feed Rules: Cutting Off the Prion Recycling Route
U.S. regulations have restricted the use of certain animal proteins in ruminant feed for decades, designed to prevent
a BSE amplification loop in cattle populations. This is one of the most important prevention strategies because it targets
the mechanism that helped BSE spread in the first place.
2) Surveillance: Looking for BSE Where It Would Show Up
USDA conducts ongoing BSE surveillance, focusing testing on higher-risk cattle populations to detect disease even at extremely low levels.
This isn’t just “checking a box”it’s the epidemiologic equivalent of turning on the lights and scanning the room.
3) Slaughter Controls: Removing Specified Risk Materials (SRMs)
Another major protection is the required removal of specified risk materials (SRMs)parts of cattle most likely to contain prions if infection existed.
In general terms, that includes certain nervous system tissues (like brain and spinal cord) and related structures, handled under strict rules.
The combined effect of feed controls, surveillance, and SRM rules is why public health agencies describe the risk of vCJD in the U.S. as very low.
(Very low is not the same as zero, but it’s a meaningful difference.)
Food Safety Questions People Actually Ask
“Should I stop eating beef?”
For most Americans, public health guidance does not suggest avoiding beef solely due to fear of vCJD.
U.S. food safety systems are designed to prevent BSE-contaminated materials from entering the human food supply.
If you have specific health anxieties or dietary concerns, talk with a clinician who can address your personal context.
“What about organ meats?”
Because prion risk (when it exists) is tied to certain tissues, regulations focus heavily on preventing high-risk materials from entering food channels.
If you’re purchasing specialty products, buy from reputable suppliers and follow any food safety advisories and recalls.
“Can cooking kill prions?”
Normal cooking temperatures that handle bacteria and viruses are not considered reliable for prions.
That’s why prevention relies on controlling what enters the food supplynot expecting your grill to do biomedical magic.
Treatment: What Can Be Done?
At present, there is no cure for CJD or vCJD, and there is no treatment proven to stop disease progression.
Care focuses on comfort, safety, and quality of life:
- managing pain, anxiety, insomnia, or agitation,
- treating muscle jerks or movement symptoms when possible,
- supporting swallowing and nutrition decisions,
- preventing falls and injuries,
- and engaging palliative care and hospice early for support.
Families often describe this as a “sudden storm” illness: everything changes fast, and support systems matter as much as medical tests.
Healthcare Infection Control: Why Prion Protocols Exist
Prions are unusual. Because they can be more resistant than typical pathogens, hospitals may use special handling and sterilization protocols
when prion disease is suspectedespecially for neurosurgical instruments and certain procedures.
If a clinician suspects prion disease, they may coordinate with infection control teams and reference laboratories.
This protects future patients and helps ensure samples and instruments are managed appropriately.
Frequently Asked Questions (FAQ)
Is classic CJD the same thing as Alzheimer’s?
No. CJD can cause dementia symptoms, but it progresses far more rapidly than typical Alzheimer’s disease.
Also, CJD is a prion disease with different biology and diagnostic testing.
Can a person survive CJD?
CJD is considered almost always fatal. Research continues, but current care focuses on support and comfort.
How rare are these diseases?
Classic CJD occurs worldwide at a rate often described around one to two cases per million people per year.
vCJD is far rarer, especially in the United States.
If someone has rapidly worsening confusion, what should we do?
Seek urgent medical evaluation. Many causes of rapidly progressive dementia are treatable (including autoimmune and infectious conditions),
and early diagnosis can change outcomes.
Conclusion
Creutzfeldt-Jakob disease and mad cow disease are linked by one eerie concept: the prion.
But “linked” doesn’t mean “common,” and it doesn’t mean “inevitable.”
Classic CJD is usually sporadic and not something you catch from day-to-day life. Variant CJD is the form historically connected to BSE,
and its rarityespecially in the U.S.reflects layers of prevention: feed controls, cattle surveillance, and strict rules around high-risk tissues.
The most important takeaway is practical: if you ever see rapid neurological decline in someone you love, treat it as a medical emergency.
The goal isn’t to self-diagnose CJDit’s to get fast, expert evaluation for conditions that might be reversible, and compassionate support if they are not.
Experiences: What Patients, Families, and Clinicians Often Share (A 500-Word Perspective)
People rarely come to prion disease stories through casual curiosity. Most arrive through a confusing, frightening moment:
someone is “not themselves,” and it’s getting worsefast. Families often describe the early phase as a whiplash mix of denial and urgency.
A loved one might start with small but unsettling changesmisplacing words, acting oddly impulsive, stumbling, or struggling with simple tasks
and within weeks the decline can feel shockingly steep. The speed is what makes prion disease emotionally different from many other dementias:
there’s little time to adjust, and the normal rhythm of “plan, adapt, cope” gets compressed into a blur.
Clinicians who evaluate rapidly progressive dementia often talk about the “diagnostic odyssey” from the medical side.
The first job is to look for treatable causesautoimmune encephalitis, unusual infections, medication effects, metabolic problems
because those can sometimes be reversed if caught early. That means lots of tests, specialist consults, and sometimes multiple hospital visits.
Families can feel like they’re living inside a medical checklist, waiting for someone to say, “Here’s the answer.”
When the possibility of CJD enters the conversation, it can land like a thunderclapespecially because there’s no cure and the word “fatal” arrives early.
A theme many caregivers mention is how prion disease affects identity and communication.
Rapid cognitive changes can lead to agitation, fear, or personality shifts that feel out of character.
Caregivers sometimes wrestle with guilt: “Should I have noticed sooner?” or “Did I do something wrong?”
The hard truth is that prion disease is not caused by ordinary mistakes, and most families could not have prevented it.
What families can control is the response: getting expert evaluation, prioritizing safety (especially fall prevention),
and building a support team quickly.
Practical needs show up immediately. People often need help with mobility, toileting, feeding decisions, and supervision.
Many families say the most helpful turning point was involving palliative care or hospice earlier than they expected.
That support is not “giving up.” It’s bringing in experts who focus on comfort, symptom relief, caregiver training,
and making the home environment safer and calmer. Even small adjustmentsnight lights, simplified routines, quiet surroundings,
and a consistent caregiver rotationcan reduce distress.
Another repeated experience is the value of community, even when the disease is rare.
Families often feel isolated because few people have heard of CJD, and friends may not understand why everything is moving so quickly.
Talking with others who have lived through it can help caregivers feel less alone, learn what to expect,
and find language for difficult conversationslike choosing comfort-focused care, planning for end-of-life needs,
or coordinating paperwork and work leave.
If there’s one “shared lesson” clinicians and families often echo, it’s this:
focus on what helps today. With prion disease, the timeline can be cruelly short, but supportive care can still reduce suffering,
preserve dignity, and help families feel grounded in the middle of chaos. Love looks less like heroic medical breakthroughs
and more like steady presence, comfort, safety, and getting help before you’re running on fumes.
![18 Best Types of Charts and Graphs for Data Visualization [+ How to Choose]](https://corkopencoffee.org/wp-content/uploads/2026/05/18-best-types-of-charts-and-graphs-for-data-visualization-how-to-choose-qKM1PBYG-thumb.jpg)




